
美國食品和藥物管理局(Food and Drug Administration)簡稱FDA,FDA 是美國政府在健康與人類服務部 (DHHS) 和公共衛生部 (PHS) 中設立的執行機構之一。作為一家科學管理機構,FDA 的職責是確保美國本國生產或進口的食品、化妝品、藥物、生物制劑、醫療設備和放射產品的安全。
美國食品藥物管理FDA于官網發布標題為“器械注冊和列名”的公告,并著重強調:
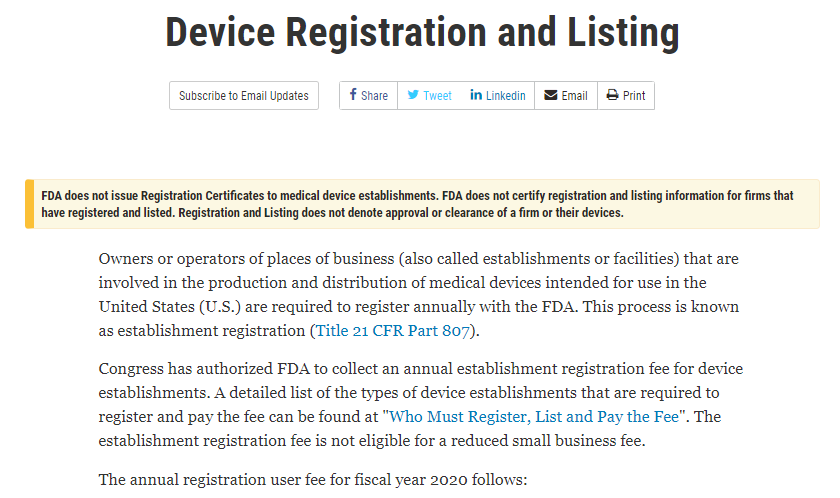
FDA does not issue Registration Certificates to medical device establishments. FDA does not certify registration and listing information for firms that have registered and listed. Registration and Listing does not denote approval or clearance of a firm or their devices.
FDA不會向醫療器械企業頒發注冊證書,不會對已經注冊或者列名的產品或企業出具確認證書。企業注冊和產品列名信息并不代表FDA批準了該企業和其產品。
FDA注冊我們一般需要關注的問題點如下:
答:FDA注冊是沒有證書的,產品通過在FDA進行注冊,將取得注冊號碼,FDA會給申請人一份回函(有FDA行政長官的簽字),但不存在FDA證書一說。
FDA在此時發布這樣一則通告可謂是強提醒!由于近期美國疫情的發展,醫用防疫用品出口美國的需求量大量增加,出口注冊需求也就增多,但是存在一些企業冒充FDA給廠家頒發證書,一些經銷企業在咨詢廠家的時候,拿到的“FDA證書”也可能是假冒的。
答:FDA是一個執法機構,而不是服務機構。如果有人說他們是FDA下屬的認證實驗室,那么他至少是在誤導消費者,因為FDA既沒有面向公眾的服務性認證機構與實驗室,也沒有所謂的“指定實驗室”。FDA作為聯邦執法機構,不可以從事這種既當裁判又當運動員的事。FDA只會對服務性的檢測實驗室的GMP質量進行認可,合格的頒發合格證書,但不會向公眾“指定”,或推薦特定的一家或幾家。
答:是的,企業在進行FDA注冊時必須指派一名美國公民(公司/社團)作為其代理人,該名代理人負責進行位于美國的過程服務,是聯系FDA與申請人的媒介。
1、FDA注冊和CE認證不同,他認證的模式不同于CE認證的產品檢測+報告證書模式,FDA注冊實際上采用的是誠信宣告模式,即:你對自己的產品符合相關標準和安全要求負責,并在美國聯邦網站注冊,如果產品出事,那么就要承擔相應的責任。因此FDA注冊對于大部分產品,不存在寄樣品檢測和出證書的說法。
2、FDA注冊有效期問題:FDA注冊有效期為一年,如果超過一年,則需要重新提交注冊,所涉及的年費也需要重新付。
3、FDA注冊是否有證書?
實際上,FDA注冊是沒有證書的,產品通過在FDA進行注冊,將取得注冊號碼,FDA會給申請人一份回函(有FDA行政長官的簽字),但不存在FDA證書一說。我們通常看到的這個證書是中介代理機構(注冊代理)簽發給廠家,以證明其幫助該廠家完成了美國FDA要求的“生產設施注冊和產品類型注冊”(Establishment Registration and Device Listing),完成的標志是幫助廠家取得了FDA的注冊登記號。

根據風險等級的不同,FDA將醫療器械分為三類(Ⅰ,Ⅱ,Ⅲ),Ⅲ類風險等級最高。
FDA將每一種醫療器械都明確規定其產品分類和管理要求,目前FDA醫療器械產品目錄中共有1700多種。任何一種醫療器械想要進入美國市場,必須首先弄清申請上市產品分類和管理要求。
在明確了以上信息后,企業就可以著手準備有關的申報資料,并按一定程序向FDA申報以獲取批準認可。對于任何產品,企業都需進行企業注冊和產品列名。
對Ⅰ類產品(占47%左右),實行的是一般控制,絕大部分產品只需進行注冊、列名和實施GMP規范,產品即可進入美國市場(其中極少數產品連GMP也豁免,極少數保留產品則需向FDA遞交510(K)申請即PMN (Premarket Notification));
對Ⅱ類產品(占46%左右),實行的是特殊控制,企業在進行注冊和列名后,還需實施GMP和遞交510(K)申請(極少產品是510(K)豁免);
對Ⅲ類產品(占7%左右),實施的是上市前許可,企業在進行注冊和列名后,須實施GMP并向FDA遞交PMA(Premarket Application)申請(部分Ⅲ類產品還是PMN)。
對Ⅰ類產品,企業向FDA遞交相關資料后,FDA只進行公告,并無相關證件發給企業;對Ⅱ、Ⅲ類器械,企業須遞交PMN或PMA,FDA在公告的同時,會給企業以正式的市場準入批準函件,即允許企業以自己的名義在美國醫療器械市場上直接銷售其產品。
至于申請過程中是否到企業進行現場GMP考核,則由FDA根據產品風險等級、管理要求和市場反饋等綜合因素決定。
綜合以上內容可知,絕大部分產品在進行企業注冊、產品列名和實施醫療器械GMP,或再遞交510(K)申請后,即可獲得FDA認證。
唯一權威途徑:上FDA官網查詢
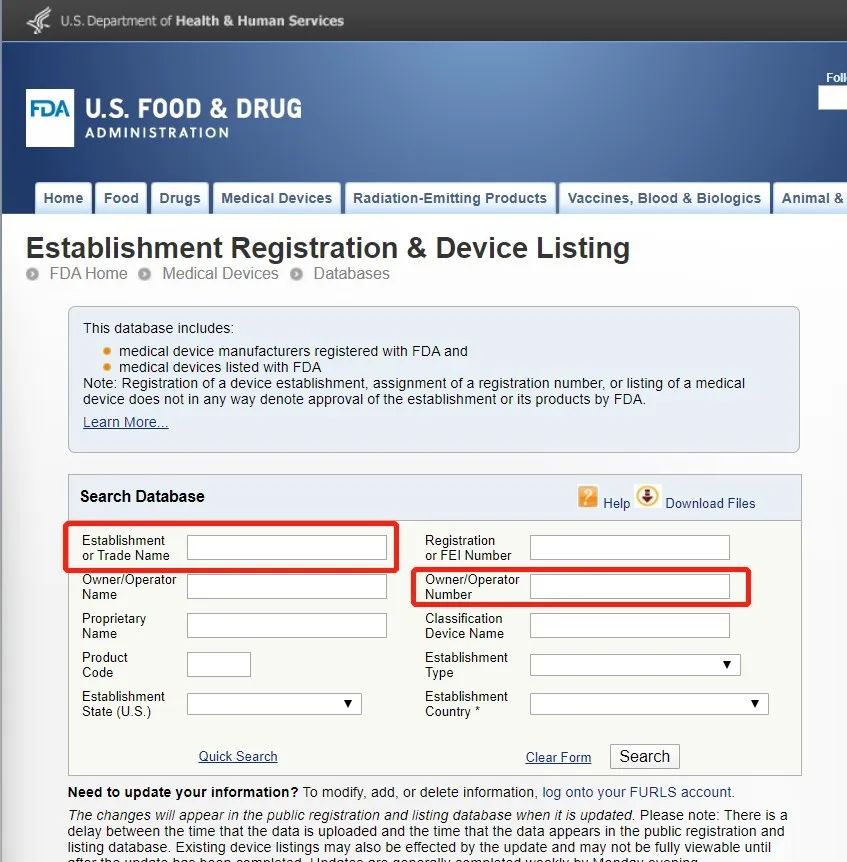
● FDA列名:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm
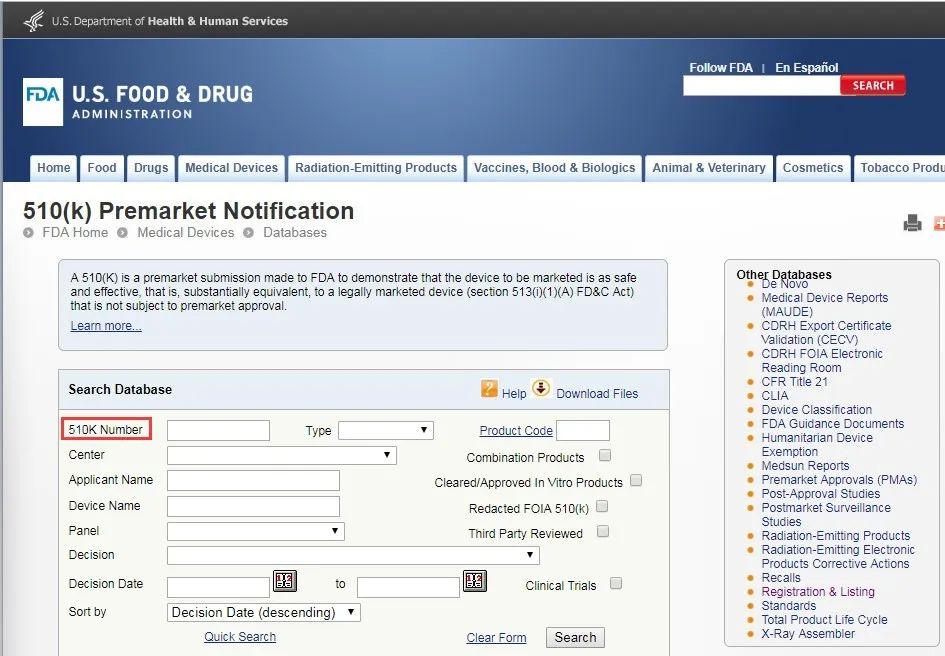
● 510K注冊:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
【來源:internet】
版本與免責申明:
①本網凡注明“來源:xxx(非本網)”的作品,均轉載自其它媒體,轉載目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,且不承擔此類作品侵權行為的直接責任及連帶責任。
②如涉及作品內容、版權等問題,請在作品發表之日起兩周內與本網聯系,我們將在第一時間刪除內容。



